Help
What is MIMP?
Mutations IMpact on Phosphorylation (MIMP) is a web service that predicts if a mutation within a phosphorylation site will alter phosphorylation. MIMP works by constructing specificity models for kinases, which are then used to score phosphosites containing a mutation before and after the mutation to predict the impact it may have on phosphorylation. There are three types of events that can occur
- Loss-of-phosphorylation: the mutation disrupts specificity for a kinase
- Gain-of-phosphorylation: the mutation creates a new motif which causes phosphorylation by a new kinase
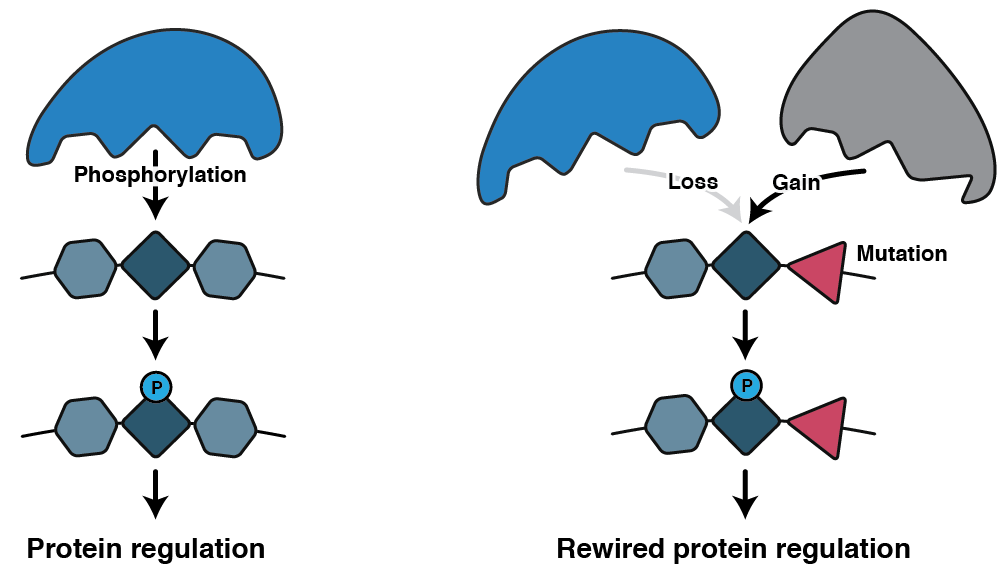
How do I use it?
To use MIMP you will need two mandatory files and one optional file.
- FASTA file of protein sequences. For example:
>seq0 FQTWEEFSRAAEKLYLADPMKVRVVLKYRHVDGNLCIKVTDDLVCLVYRTDQAQDVKKIEKF >seq1 KYRTWEEFTRAAEKLYQADPMKVRVVLKYRHCDGNLCIKVTDDVVCLLYRTDQAQDVKKIEKFHSQLMRLME LKVTDNKECLKFKTDQAQEAKKMEKLNNIFFTLM
Note: Avoid using spaces in your fasta headers. If spaces are used, MIMP will use the text before the first space as the header. For example,
>seq more textwill become>seq. If you do use spaces in your headers, make sure your mutation and phosphorylation data correspond to the equivalent of>seqin this case. - Mutation data. This should contain the name of the protein and the mutation, space delimited. The mutation should have a
X123Zformat, whereXis the reference amino acid,Zis the alternate amino acid and123is the position of the substitution. Protein names should correspond to that of the fasta file. For example:seq0 F1H seq1 T4V seq1 W5L
- Phosphorylation site data (optional but recommended). This should contain the name of the protein and the position of phosphoacceptor S, T or Y residue, space delimited. Protein names should correspond to that of the fasta file. For example:
seq0 8 seq0 15 seq1 9
All positions must correspond to an S, T or Y residue. If you are not providing phosphorylation site data, MIMP will consider each S, T or Y residue as a potential phosphoacceptor.
What are the different kinase specificity models?
There are three different sets of specificity models. The chosen set is used in the scanning of phosphorylation rewiring in MIMP.
- Individual kinase specificity models these are specificity models constructed using experimentally verified kinase-peptide phosphorylation on an individual kinase level. Data is obtained from public databases such as PhosphoSitePlus, PhosphoELM and HPRD. Since these models are constructed using experimental data on the peptide level, they are considered high confidence.
- Kinase family specificity models This is similar to 1. Since kinase families typically have similar specificities, these models are also constructed using experimentally verified kinase-peptide phosphorylation, but are grouped based on the kinase families/subfamilies. This can have certain advantages and disadvantages. For example, there are cases where members of kinase families show a moderate and sometimes large difference from that of the collapsed family model. However, using kinase family models can avoid redundant (gain or loss of phosphorylation) prediction of family members that have similar specificities.
- Predicted individual kinase specificity models these are specificity models that are predicted from kinase-substrate interactions, on the protein level and not the peptide. The kinase-substrate relationships (KSRs) are derived using protein microarrays as described in Newman et al. (2013). The KSRs are then combined with experimentally identified phosphosites to predict the specificity models by through an iterative refinement process (termed M3) similar to that described in the MIMP paper. For more information on the M3 algorithm please see the Newman et al. (2013) paper.
Please be extremely cautious when using these models. While KSRs are identified in vitro and some models may be correct, it is likely that others are not. These models are thus considered low-confidence and we strongly urge users who use this dataset to manually verify that the logos appear valid before proceeding with downstream analyses.
What do the scores mean?
The scores quantitatively represent how well a phosphosite is likely to be phosphorylated by a kinase. Scores range from 0-1, where 0 represents no predicted phosphorylation and 1 represents perfect phosphorylation.
Peptides are automatically given a score of 0 if they have a central residue which does not match the class of the specificity model it is being scored against. There are two classes of specifcity models: serine/threonine models and tyrosine models. For example, if the tyrosine-centered phosphosite HAWNKDRYQIAICPN is scored against a serine/threonine model, it will always have a score of 0. If a peptide is mutated at the central residue to a non-STY residue, e.g. HAWNKDRAQIAICPN it will always have a score of 0.
What is the probability threshold?
Once a score is obtained for both the wild type and mutant phosphosite, MIMP will use kinase specificity models to score phosphosites that belong to a kinase (positive) and that do not (negative). This results in two distributions being generated: foreground (FG) and background (BG). MIMP then computes a probability of the wild type or mutant score falling under either one of those distributions.
The probability of a phosphorylation loss is computed as the joint probability of the wild type score falling under the FG and the mutant score falling under the BG. Similarly, the probability of a phosphorylation gain is computed as the joint probability of the wild type score falling under the BG and the mutant score falling under the FG.
Since the joint probability is computed as the product of the individual probabilities, the minimum probablity required for a phosphorylation loss or gain to be called is 0.5. Any events with gain or loss probabilities less than the probability threshold are discarded. The threshold of 0.5 should suffice for calling gains and losses. However, you may wish to increase this threshold to display only events of higher confidence.
What does the log ratio mean?
The log ratio measures how distant the wild-type score is from the mutant. A high positive log ratio represents a high confidence gain-of-phosphorylation event. A high negative log ratio represents a high confidence loss-of-phosphorylation event.
Events with a log ratio below the log ratio threshold are discarded. By default, this threshold is 1, but can be increased for in the options panel.
Note: If the log ratio has a value of -, this means a log ratio cannot be assigned due to the fact that one of the scores has a value of 0. You can assume this log ratio is of high confidence i.e. approaches infinity (for gains) or -infinity (for losses)
Can I use MIMP offline?
Yes. MIMP is freely available as an R package, which can be run locally. You can find more information on how to obtain the R package on the download page
I have a large amount of data, how can I speed up the process?
If you are processing a large amount of data, please download and run MIMP locally. Please refer to the download page for more information on how to download MIMP.
I can't find my kinase of interest, what should I do?
MIMP uses up-to-date data from PhosphoELM, PhosphoSite Plus, HPRD and PhosphoNetwork. If your kinase is not available it is likely that there is insufficient kinase-substrate data available to construct a specificity model. If you are able to provide sufficient kinase-substrate data for your kinase, please contact us.
I'm having trouble with MIMP that isn't addressed on this page. What should I do?
If you are having trouble with MIMP please contact us and we will try resolve it.